WGS基础流程
本文内容:
- WGS基础流程
- WGS文件流
- WGS文件格式
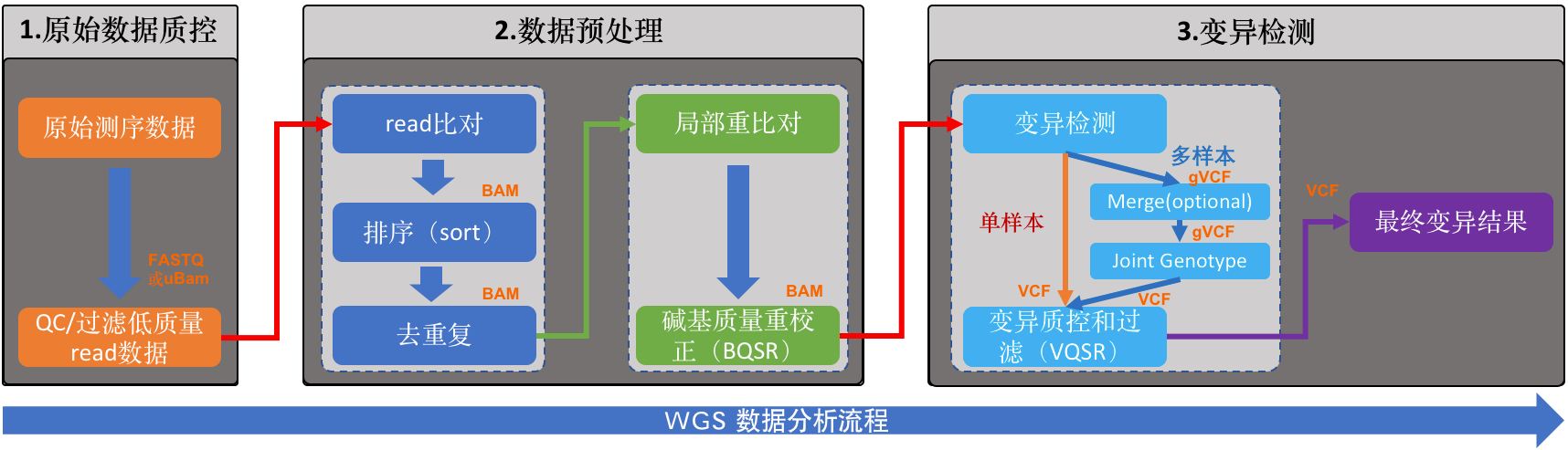
WGS基础流程
大体上分为三步:
- 原始数据质控
- 数据预处理
- 变异检测
具体一些的流程参考下面的流程图
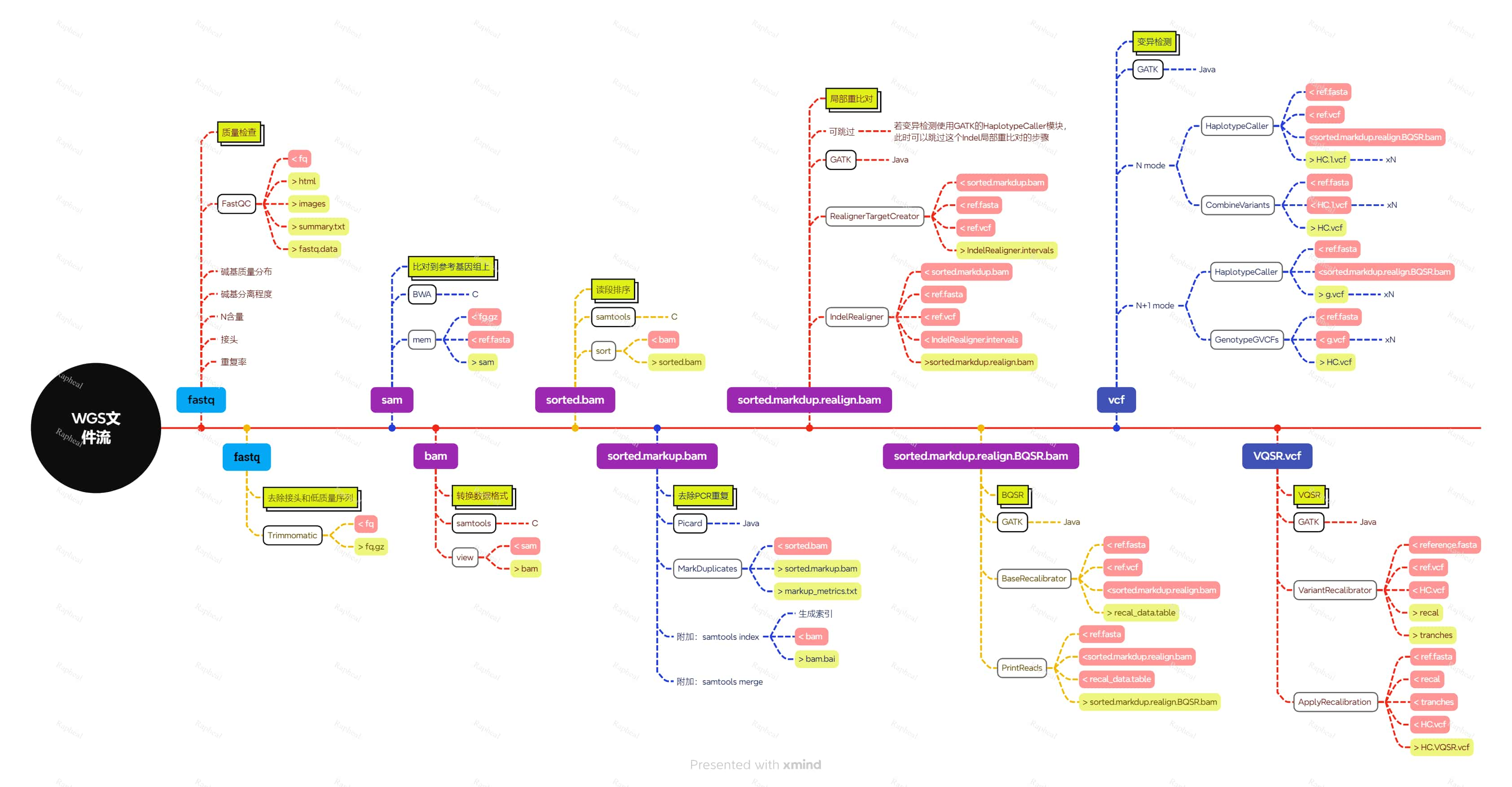
WGS文件流
我自己整理了一个思维导图:
- 荧光黄加双方框:步骤名称
- 淡红色方块:输入文件
- 荧光黄方块:输出文件
- 黑色方框:软件
- 灰色方框:软件的模块
WGS文件格式
本节介绍的文件格式:
- fastq
- fasta
- bam & sam & cram & bai
- vcf
fastq
1 | @DJB775P1:248:D0MDGACXX:7:1202:12362:49613 |
类型:文本文件
内容:产生自测序仪的原始测序数据
标识:.fastq .fq .fq.gz
组成:每四行成为一个独立的单元,我们称之为read。
- 第一行:以‘@’开头,是这一条read的名字,这个字符串是根据测序时的状态信息转换过来的,中间不会有空格,它是每一条read的唯一标识符,同一份FASTQ文件中不会重复出现,甚至不同的FASTQ文件里也不会有重复;
- 第二行:测序read的序列,由A,C,G,T和N这五种字母构成,这也是我们真正关心的DNA序列,N代表的是测序时那些无法被识别出来的碱基;
- 第三行:以‘+’开头,在旧版的FASTQ文件中会直接重复第一行的信息,但现在一般什么也不加(节省存储空间);
- 第四行:测序read的质量值,这个和第二行的碱基信息一样重要,它描述的是每个测序碱基的可靠程度,用ASCII码表示。
质量值:碱基测量错误率越低,质量值就越高。测序错误率为p_error,质量值为Q:Q = -10lg(p_error)
ASCII码与质量值Q的对应关系标准如下所示,我们把加33的的质量值体系称之为Phred33,现在一般都是使用Phred33这个体系
| 测序平台 | ASCII码范围 | 下限 | 质量值类型 | 质量值范围 | 备注 |
|---|---|---|---|---|---|
| Sanger, Illumina(版本1.8及以上) | 33-126 | 33 | Phred quality score | 0-93 | 现在沿用 |
| Solexa, Illumina早期版本(<1.3) | 59-126 | 64 | Solexa quality score | 5-62 | 除了已测序数据之外,不再使用 |
| Illumina(版本1.3-1.7) | 64-126 | 64 | Phred quality score | 0-62 | 除了已测序数据之外,不再使用 |
fasta
1 | >gene_00284728 length=231;type=dna |
类型:文本文件
内容:存储着有顺序的序列数据
(如参考基因组序列、蛋白质序列、编码DNA序列、转录本序列)
标识:.fasta .fa .fa.gz
组成:
- 头信息:一个空格把头信息分为两个部分。第一部分是序列名字,它和大于号(>)紧接在一起;第二部分是注释信息,这个可以没有,就看具体需要。
- 序列:具体的序列数据
bam & sam & cram & bai
sam是一个存储着序列比对数据的文本文件;bam是其二进制压缩格式,也是目前通用的对比数据存储格式;cram是bam的高压缩格式;bai是bam文件的索引文件
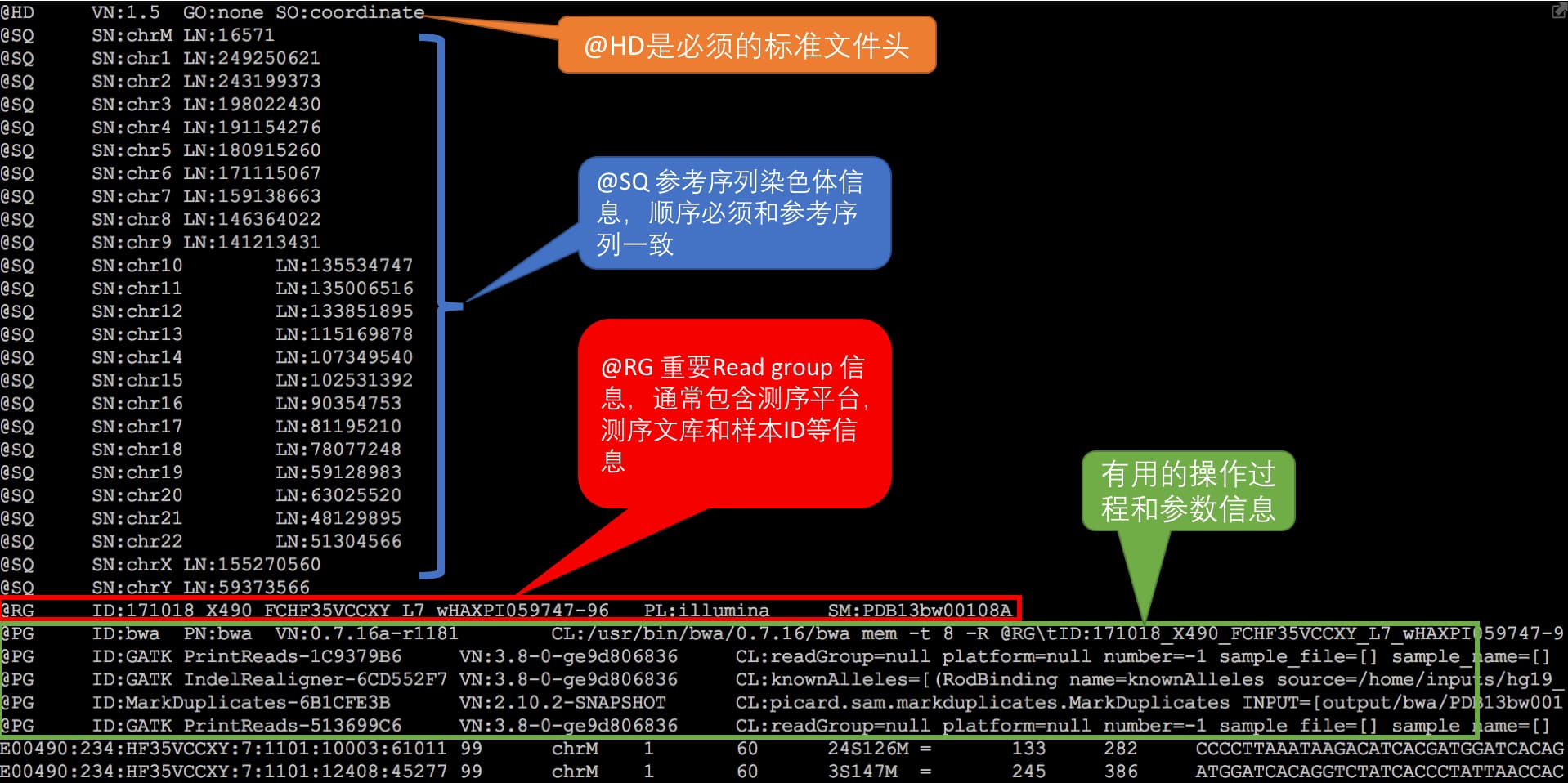
组成:SAM文件分为两个部分:header和record。这里额外说一句,许多NGS组学数据的存储格式都是由header和record两部分组成的。
- header内容不多,也不会太复杂,每一行都用‘@’ 符号开头,里面主要包含了版本信息,序列比对的参考序列信息,如果是标准工具(bwa,bowtie,picard)生成的BAM,一般还会包含生成该份文件的参数信息(如上图),@PG标签开头的那些。
- 这里需要重点提一下的是header中的@RG也就是Read group信息,这是在做后续数据分析时专门用于区分不同样本的重要信息。它的重要性还体现在,如果原来样本的测序深度比较深,一般会按照不同的lane分开比对,最后再合并在一起,那么这个时候你会在这个BAM文件中看到有多个RG,里面记录了不同的lane,甚至测序文库的信息,唯一不变的一定是SM的sample信息,这样合并后才能正确处理。
- record:这是我们通常所说的序列比对内容,每一行都是一条read比对信息,recoed中的每一个信息都是用制表符tab分开的,如下图所示:
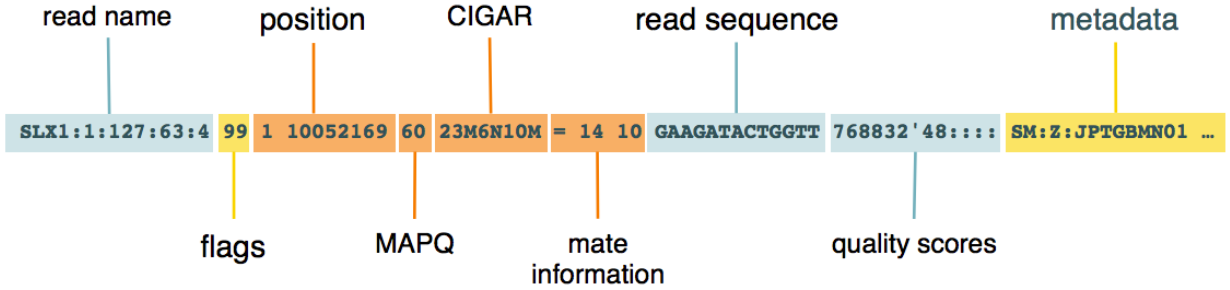
| 对应上图 | 列序号 | 名称 | 内容描述 |
|---|---|---|---|
| read name | 1 | QNAME | fq的read ID (QNAME中的Q表示Query) |
| flags | 2 | FLAG | 比对信息位 (bitwise FLAG),是一个由16位整数 |
| position | 3 | RNAME | 参考序列染色体名称 (RNAME中的R表示Reference) |
| position | 4 | POS | 比对位置,从对应染色体的第1位开始往后计算 |
| MAPQ | 5 | MAPQ | 比对质量值 (Mapping Quality) |
| CIGAR | 6 | CIGAR | 比对信息 |
| Mate information | 7 | RNEXT | 配对read所比对到的染色体 (仅Pair end 测序的数据才有) |
| Mate information | 8 | PNEXT | 配对read所比对到的位置 (仅Pair end 测序的数据才有) |
| Mate information | 9 | TLEN | 插入片段长度 (仅Pair end 测序的数据才有) |
| Read sequence | 10 | SEQ | read序列 |
| Quality scores | 11 | QUAL | read质量值 |
| metadata | 12 | Metadata | 元信息,从第12列开始往后都是metadata,一般会包括RG信息, missmatch信息等 |
| … | … | … | … |
vcf
vcf (Variant Call Format)是一种用于存储基因组序列中的变异信息
- 一般用在 单核苷酸变异(SNV),小片段插入缺失(INDEL)等
- 也用于 拷贝数变异(CNV),SV(结构变异)等
- SNV:参考基因组在1号染色体7845190为 C,但检测样本在同样位置为 A
- INDEL:包含插入和缺失两种
- Insertion:参考基因组某片段为 ACTTG,但是检测样本同样位置为 ACCCTTG,插入了CC
- Deletion:参考基因组某片段为 TTCGG,但是检测样本同样位置为 TTGG,缺失 C
1 | #CHROM POS ID REF ALT QUAL FILTER INFO |
组成:文件一般包含两部分,
- 注释信息(header):位于文件开始,每行以 #开始
- 变异信息(body):没有 #即为记录的变异信息
| 字段 | 描述 | 举例 |
|---|---|---|
| CHROM | 染色体号,注意不需要前缀 chr | 1 |
| POS | 变异位点,对于 INDEL,为 INDEL 的第一个碱基位点 | 10616 |
| ID | dbSNP 的编号,.为置空 | rs376342519 |
| REF | 参考基因组的碱基,也就是等位基因 | CCGCCGTTGCAAAGGCGCGCCG |
| ALT | 检测样本的碱基,同一位置有多个则以,分隔 | C |
| QUAL | Phred 的质量值,表示改位点存在变异的可能性。分数越高则认为越可靠,但同时需要考虑测序深度,覆盖度等因素。.代表置空,不代表质量值为 0。 | 100 |
| FILTER | 过滤标志,如果为 PASS则认为是一个变异 | PASS |
| INFO | 详细信息,用 key=value的格式来表示。key 一般是简写,具体描述在文件开头的 header lines 中显示。 | AC=4973;AF=0.993011;AN=5008;VT=INDEL |
| FORMAT | 可选,变异位点格式,包括 GT,AD,DP,GQ,PL/ GT,AD,DP,GQ,PGT,PID,PL,PS | GT:DP:GQ:PL |
| SAMPLEs | 可选,各个样本的值,来自 BAM 文件 @RG 的 SM 标签。一般每个样本对应一列,因此该文件会超过十列。每个样本会与 FORMAT 列的格式一一对应,不同格式用 :分隔 | 0/1:50:99:0,20,200 |
这部分暂时咕咕
WGS软件
本节介绍的软件:
- FastQC
- Trimmomatic
- BWA
- Samtools
- Picard
- GATK

