孟德尔随机化笔记
这是一份双样本孟德尔随机化(MR)的代码实操笔记,在这里你可以了解如何用R,在自己的电脑上判断一下两个表型之间是否具有潜在的因果关系。因为是刚刚接触MR,难免有不准确之处,本文仅作参考。
1,获取数据
现存 GWAS 数据样本大多为欧洲样本,少量其他样本
关键要点:暴露与结局数据不存在或存在少量可接受范围内的样本重叠
必须数据(
snp||p||effect_allele||other_allele||or(beta)||se)SNP_ID (rs123456):唯一识别号
p_value:snp 位点与表型的相关程度( 0 < p < 1)
effect_allele:变异位点
other_allele:常规位点
Beta:连续型变量的相对风险比,(R,大于 0 促进发生,小于 0 抑制表型)
OR:二分类变量的相对风险比,(OR > 0,大于 1 促进发生,小于 1 抑制表型)
Beta = ln(OR)
Standard error(se):标准差,代表相对风险比 Beta 的准确性
(非必须)Effect allele frequency(eaf):效应位点频率,算工具变量的时候需要,计算 R^2 时需要 MAF
2,读取数据
首先设置工作环境,R-> NewProject-> working Env,文件名可以有路径
更快的数据读取方法:
data.table包下的fread()函数可替代 read.table()“snp_gwas.tbl.txt” -> 直接用
read.table()读取,可加header = T- .txt :文本文件,可以用记事本打开的
1
2data <- read.table("snp_gwas.tbl.txt",header = T)
View(data)“snp_gwas.txt.gz” -> 直接用
read.table()读取,可加header = T- .gz : gzip 压缩后的文件,解压之后再读取似乎会快一点,当然也可以直接读取
1
2data <- read.table("snp_gwas.txt.gz",header = T)
View(data)“snp_gwas.xlsx” -> 可以使用
readxl包下的read_excel()函数读取1
2library(readxl)
data <- read_excel("snp_gwas.xlsx")“ieu-b-4979.vcf.gz” -> 利用
VariantAnnotation和gwasvcf包将 vcf 格式文件转换成 tibble 格式文件,函数分别对应readVcf()和vcf_to_tibble(),后者 id 应该可以不加VCF: Variant Call Format,变异调用格式,是一种常用于存储基因组变异信息的文本文件格式。它是生物信息学领域中一种标准格式,用于描述个体基因组的单核苷酸变异(SNP,Single Nucleotide Polymorphism)、插入或删除等变异信息
VCF 文件中的列描述行(Column Header Line)包含对列名的解释,这有助于理解每个列中存储的信息,列描述行位于以 #CHROM 开始的行之前,以 ##FORMAT 开始
可在 bash 命令行中使用
head -n 20 your_vcf_file.vcf查看前 20 行数据可在 R 中使用
readLines("./ieu-a-2.vcf",n=20)读取前 20 行数据可在 powershell 命令行中使用
Get-Content -TotalCount 20 your_vcf_file.vcf查看前 20 行数据1
2data0=VariantAnnotation::readVcf("ieu-b-4979.vcf.gz")
data1=gwasvcf::vcf_to_tibble(data0,id='ieu-b-4979') # 这样就能得到tibble格式的数据了,但是没有列名解释; id参数大概可以不要1
2vcf20 <- readLines("./MR数据/bmi-2/ieu-a-2.vcf",n=20)
print(vcf20)# 可以看到前20行数据1
2
3
4
5
6
7PS D:\A-MR\MR数据\bmi-2> Get-Content -TotalCount 20 ieu-a-2.vcf
##fileformat=VCFv4.2
##FILTER=<ID=PASS,Description="All filters passed">
##INFO=<ID=AF,Number=A,Type=Float,Description="Allele Frequency">
##FORMAT=<ID=ES,Number=A,Type=Float,Description="Effect size estimate relative to the alternative allele">
##FORMAT=<ID=SE,Number=A,Type=Float,Description="Standard error of effect size estimate">
(数据的格式)
3,将数据写入文件
csv:一种以逗号为分隔符的文件格式,支持 excel 打开,适合存储表格数据
使用函数
write.csv(),前一个变量是变量名,后一个 file = “文件名”1
write.csv(data,file = "exposure.csv")
4,Exposure Data 处理,获得工具变量
相关性设置
筛选条件:p < 5e-08 ( 视条件而定 )
使用 base 包的
subset( )即可1
2
3data_subseted <- subset(data,p < 5e-08)
View(data_subseted)
write.csv(data_subseted,file = "data_subseted.csv")
独立性设置
首先将 subseted 的文件放在 TwoSampleMR 文件夹下
筛选条件:LD 连锁不平衡,r^2 < 0.001(最大 0.1) , kb = 10000
读取文件路径,使用
system.file()函数,前一个参数是包下的相对路径,后一个参数是包名,主要作用是根据指定的包和文件路径获取完整的文件路径1
road <- system.file("package_folder/exposure.csv",package = "TwoSampleMR")
规范读取数据:利用 TwoSampleMR 包的
read_exposure_data()规范读取数据,参数分为四个部分:文件路径、分隔符、规范列名、最后一个是当有多个暴露时,将多个暴露合在一起跑,可以通过 phenotype_col 识别每一个暴露的标识,从而单独跑每一个暴露1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18exposure_data <- read_exposure_data(
filename = road,
sep = ",",
snp_col = "SNP_col_name",
pval_col = "p_col_name",
effect_allele_col = "effct_allele_col_name",
other_allele_col = "other_allele_col_name",
beta_col = "beta_col_name",
se_col = "se_col_name",
eaf_col = "eaf_col_name",
clump = F , ## 若为T则在读取的同时进行clump操作
phenotype_col = "exposure_type_col_name"
)独立性设置(clump): 去除连锁不平衡的 snp,利用 TwoSampleMR 包的
clump_data(),必须参数是上一步读取的数据 exposure_data,可设置参数是 clump_kb = 10000,clump_r2 = 0.0011
exposure_data_clumped <- clump_data(exposure_data)
统计强度设置
F 值可放到后面计算,影响不大
F = R^2(N-2)/(1- R^2) (F > 10 的 snp 才强相关,小于 10 的可以手动剔除)
5,Outcome Data 处理,提取结局中的 snp
导入结局数据
可能出现暴露和结局的效应等位基因不一致的情况,正常现象
可能出现暴露的部分 snp 在结局中没有,数量小可以容忍,数量大建议放弃
丢失的 snp 可以通过找代理 snp 解决,但较麻烦
1
2data <- read.table("outcome.txt",header = T)
## 如是其他文件格式,参见 “2,读取数据”
merge,提取交集,得到 outcome 数据,第一个参数是 clump 过的暴露数据名,第二个参数是刚读取的结局数据名,后面 by.x 和 by.y 分别是暴露和结局的 snp 的 rs 编号的列名,得到的数据前一部分是暴露的,后一部分是结局的,all.x 表示把暴露的 snp 的全部都找出来,在 outcome 中没有的以 NA 填充
- 当 merge 后有丢失的 snp,可以在代理 snp 网站上去找(也可以不找),一个 snp 会给你一个及以上的代理 snp,拿这些 snp 与 outcome 再做一次 merge,找出可用的 snp,对于同一个丢失 snp 的对应 snp,应当选择 R^2 最大的代理 snp 作为代理 -> 找到之后不用换 snp 的 rs 号,直接将数据填充 NA 即可
1
2
3
4
5
6
7
8
9outcome <- merge(
exposure_data_clumped,
data,
by.x = "exposure_snp_col_name",
by.y = "data_snp_col_name",
all.x = T
)
write.csv(outcome,file = "outcome.csv" )规范读取 outcome 数据,使用 TwoSampleMR 的
read_outcome_data()函数,第一个参数是暴露挑选出的工具变量,第二个参数是结局文件路径,第三个参数是间隔符,第四部分是列名1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17outcome_data <- read_outcome_data(
snps =exposure_data$SNP,
filename = "outcome.csv",
sep = ",",
snp_col = "SNP_col_name",
pval_col = "p_col_name",
effect_allele_col = "effct_allele_col_name",
other_allele_col = "other_allele_col_name",
beta_col = "beta_col_name",
se_col = "se_col_name",
eaf_col = "eaf_col_name",
)常见问题
样本重叠,用 meta 分析的暴露和结局下的队列样本数分别除以暴露和结局的 snp 数,得到的比重取最大值(具体待细查),蔡老师认为在 10%以下都可以接受
结局的 snp 数量要足够多,merge 才能丢失最少的 snp,即结局的中的 snp 多到涵盖大部分工具变量 snp
6,Harmonize Data 协同数据
Harmonize 协同作用
将 exposure_snp 和 outcome_snp 等位基因方向协同
根据 eaf 大小,剔除不能判断方向的 palindromic snp
剔除 incompatible snp (A/G vs. A/C)
使用
harmonise_data()即可,结果中会把 palindromic snp 和 incompatible snp 列出来并在 mr_keep 列标记为”FALSE”,意为在后面的 mr 分析中没有把这些 snp 纳入分析注意要保存协同后的数据
1
2
3
4
5
6dat_harmon <- harmonise_data(
exposure_dat = exposure_dat_name,
outcome_dat = outcome_dat_name
)
View(dat_harmon)
write.csv(dat_harmon,file = "dat_harmon.csv")
7,MR 分析,获得因果关系结果
可使用
mr()直接处理协同后的数据,默认使用五种方法分析(下为方法在函数中的命名)- mr_egger_regression
- mr_weighted_median
- mr_ivw
- mr_simple_mode
- mr_weighted_mode
1
dat_harmon_mr <- mr(dat_harmon)
也可使用
mr_method_list()看函数有哪些方法,并指定使用哪些方法1
2
3
4
5
6
7
8dat_harmon_mr <- mr(
dat_harmon,
method_list = c(
"mr_ivw",
"mr_egger_regression",
"mr_weighted_median"
)
)如何看待 mr 的结果
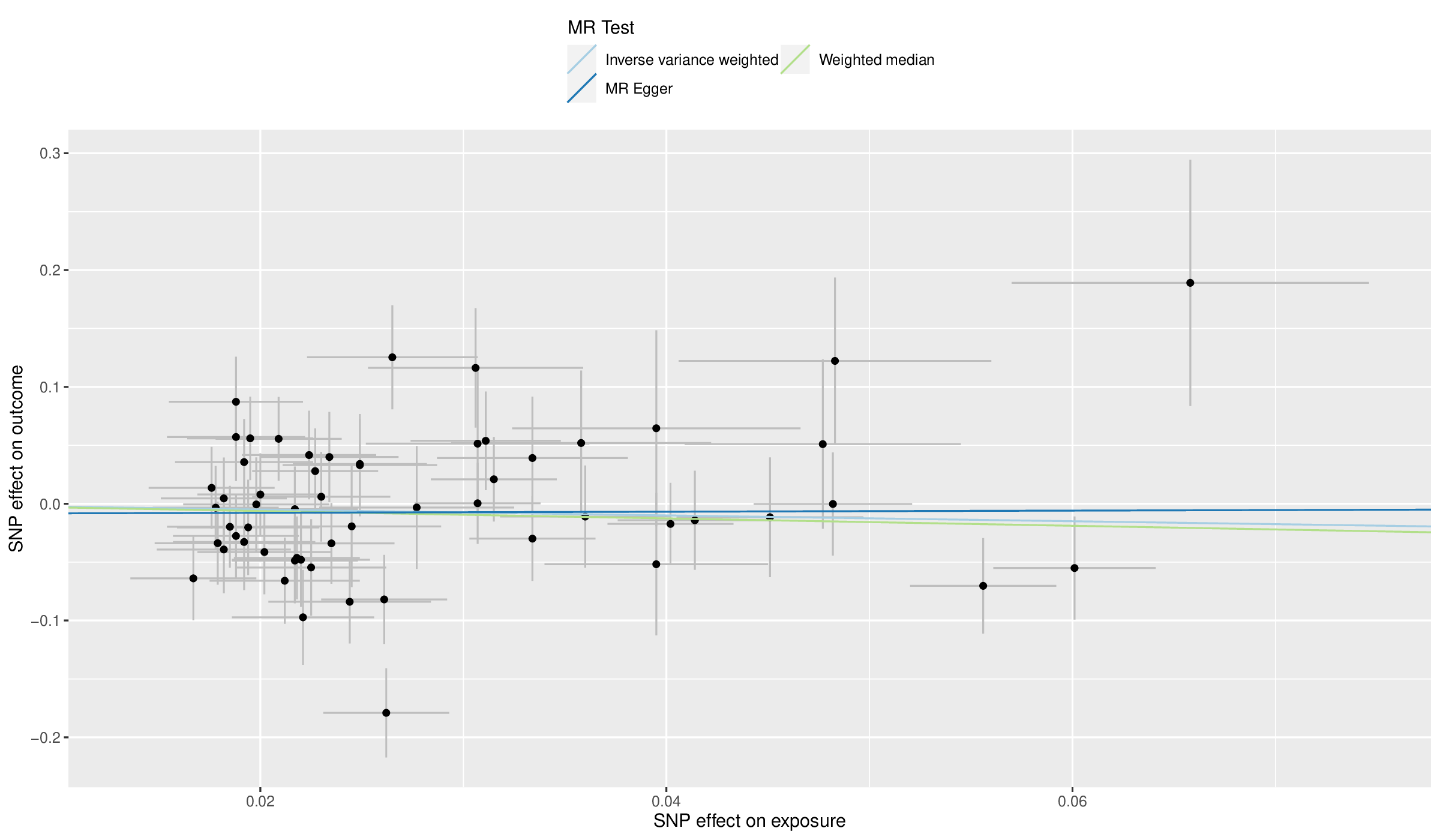
- 首先看 ivw 的 p 值,若小于 0.05 说明有因果关系
- 其次看其他方法的 beta 值,若都大于 0 或小于 0,即方向一致,则没有问题
- 若非如此,需要具体问题具体分析,有时(当 ivw 的结果不可靠时)会以 weight_median 的 p 值为准
- 没有问题就可以进入下一步分析异质性
mr 分析可视化:使用
mr_scatter_plot(),第一个参数是 mr 指定三种方法的分析结果,第二个参数是协同化的数据名1
2
3
4
5
6
7
8
9
10mr_scatter_plot(
mr_results = mr(
dat_harmon,
method_list = c("mr_ivw",
"mr_weighted_median",
"mr_egger_regression"
)
),
dat_harmon
)
8,异质性检测,
异质性检测
异质性:在 Mendelian 随机化(MR)研究中,异质性测试用于检查基因对因果效应的影响是否在不同基因位点之间存在显著差异。这有助于确定基因对因果关系的一致性
使用
mr_heterogeneity()即可得到主要看 ivw 的 Q_pval ,叫做 Cochran Q test,小于 0.05 说明有异质性
有异质性说明结果不太靠谱,但在 mr 分析中,异质性对结果的可靠性影响不大
1
dat_harmon_hetero <- mr_heterogeneity(dat_harmon)

MR_PRESSO(若存在异质性)
若要看产生异质性的 snp 是哪些,可以用 MR PRESSO,直接使用 TwoSampleMR 包下的
run_mr_presso()函数,第一个参数是协同化的数据,第二个参数相当于迭代次数,snp 越多,数字要设置的越大,跑的深度越深,算出来的离群值越准确,老师推荐 10000结果中会对每一个 snp 计算 p 值,小于 0.05 说明这个 snp 是离群值,Distortion Test $Outliers Indices 会指明是哪个 snp 的 p 小于 0.05
结果中有一个 Distortion Test $Pvalue ,大于 0.05 说明离群值没有影响,小于 0.05 说明这个离群值对结果的方向有影响,即对 SNP 的 beta 或 OR 值的一致性有影响
挑选出离群值之后,可以剔除离群值,再次进行 MR 分析
- 实例####
1
run_mr_presso(alzheimer_and_bmi_harmon,NbDistribution = 3000)
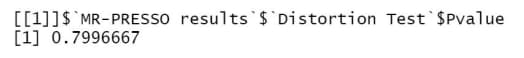
异质性可视化
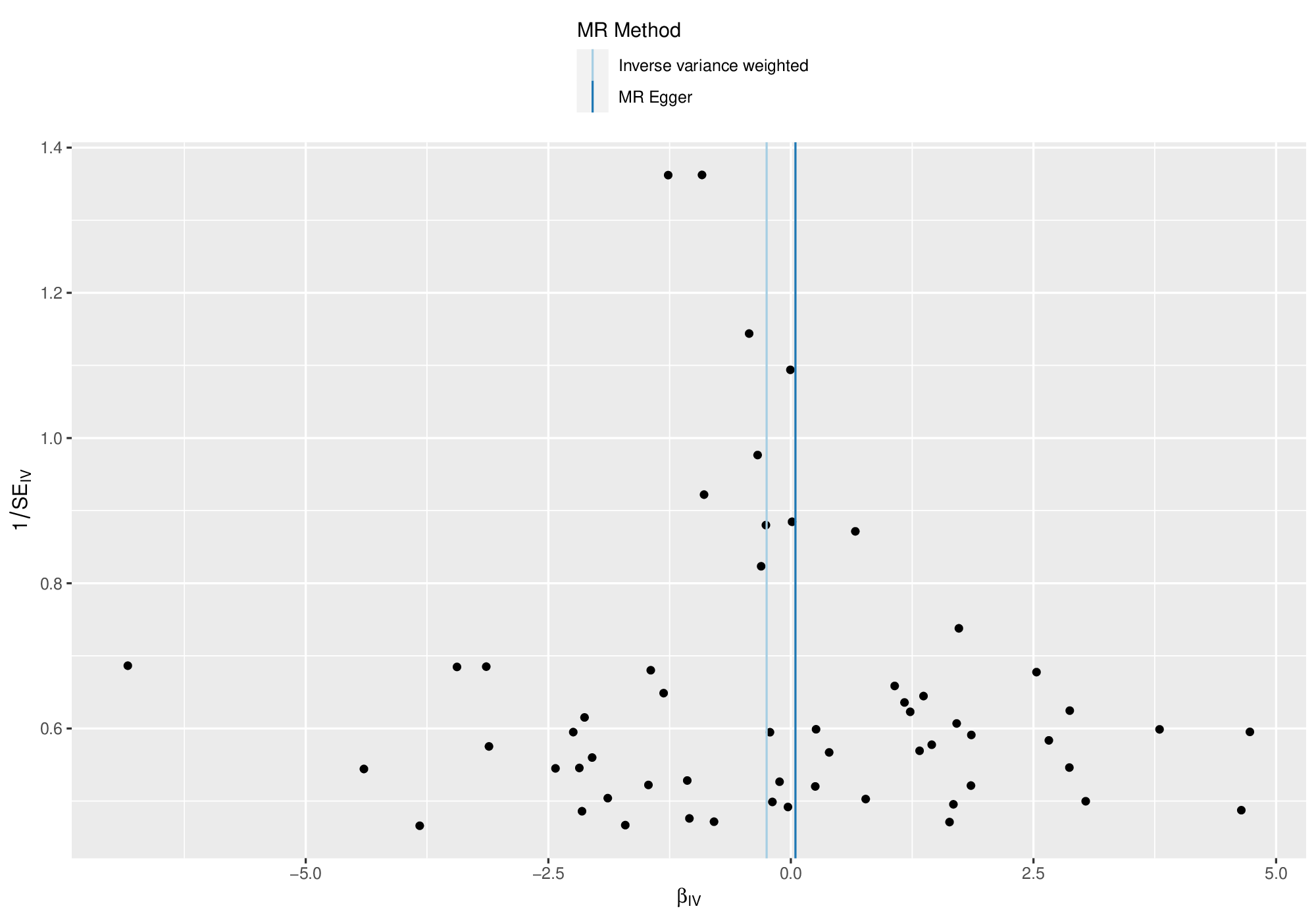
使用
mr_funnel_plot(),只有一个参数 singlesnp_results,需要使用mr_singlesnp()函数处理协同化的数据,这样即可得到漏斗图,越对称越好有两条线,ivw 和 mr_egger,在实践中,这两个漏斗图都应该对称,因为对称漏斗图表明 MR 方法的假设没有受到系统性的偏倚或异质性的影响
1
2
3mr_funnel_plot(singlesnp_results =
mr_singlesnp(dat_harmon)
)
9,多效性检测
多效性:多效性(Pleiotropy)是遗传学中的一个现象,指的是一个基因或基因变异影响多个表型特征,这些特征看似无关。这与“一个基因,一个特征”的原则相对立。多效性主要分为水平多效性和方向多效性两种类型,水平多效性代表特征之间没有强相关,方向多效性代表特征之间有强的关联
使用
mr_pleiotropy_test()函数,唯一参数为协同化的数据,当结果的 pval < 0.05 时说明有多效性,大于 0.05 时说明没有多效性,取决于 egger_intercept 即 egger 分析的回归直线的截距,意为暴露效应为 0 时结局的效应值,小于 0 对应 pval < 0.05 ,等于 0 对应 pval > 0.05多效性 p 值应当大于 0.05,没有多效性;若存在多效性,则结果不可靠
1
mr_pleiotropy_test(dat_harmon)

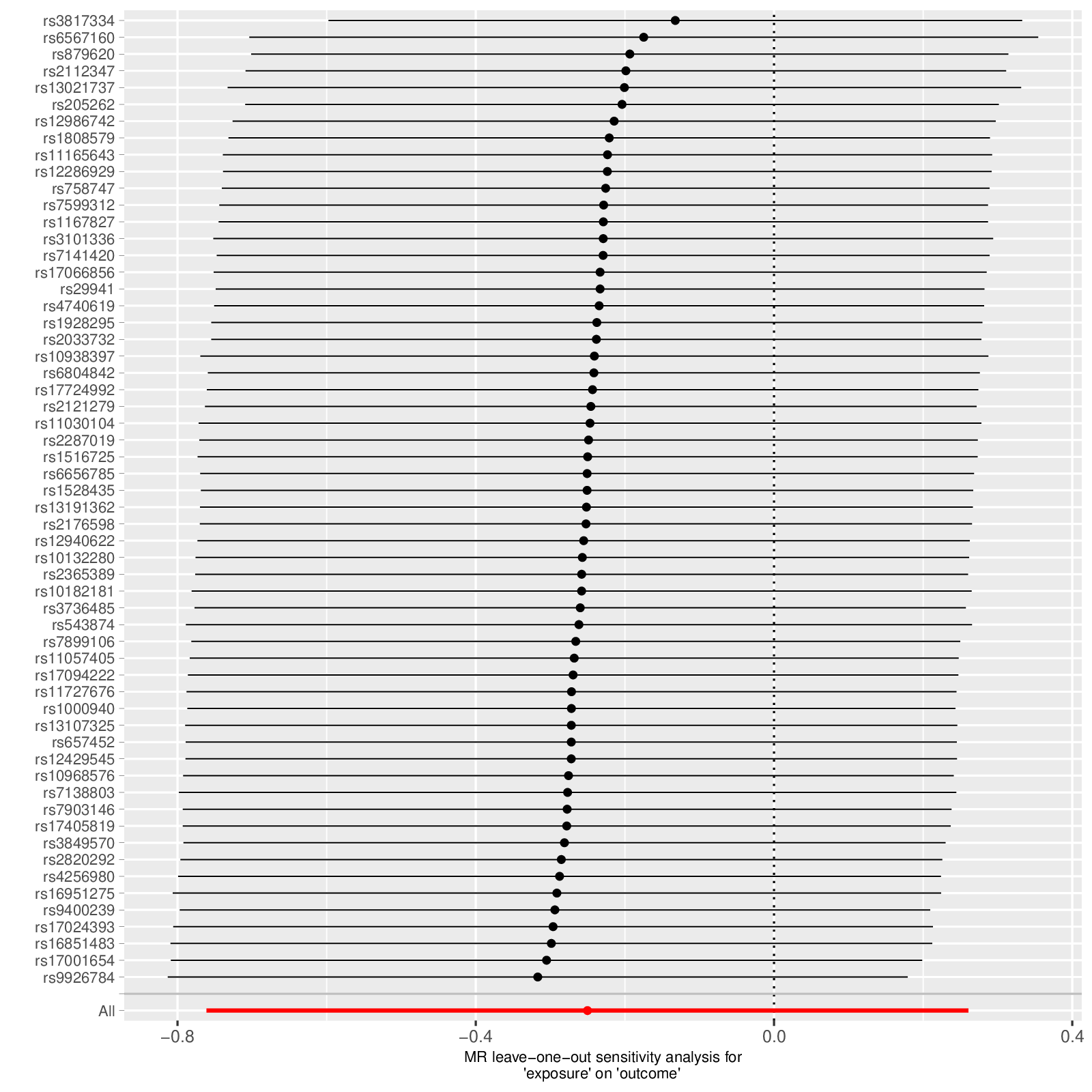
10,leave one out analysis
丢掉一个 snp 之后再次进行 mr 分析,如果是阳性结果,数据点应当都在 y 轴虚线的一侧,有人要求更加严格,需要置信区间也都在 y 轴的一侧
如果是阴性结果,则数据应当跨越 y 轴
snp 数量较少时效果比较大,snp 数量多时效果不大,但是为了研究的完整性,还是要做
使用函数
mr_leaveoneout_plot()和mr_leaveoneout(),数据为协同化的数据1
2
3
4mr_leaveoneout_plot(
leaveoneout_results =
mr_leaveoneout(dat_harmon)
)
补充:在线数据分析
在线提取数据,优点是方便,缺点是数据不够新
下面代码操作之后可以直接进入 harmonization 协同化
1
2
3
4
5
6
7
8
9
10exposure <- extract_instruments(
outcomes = "ieu-b-40"
)
View(exposure)
outcome <-extract_outcome_data(
snps = exposure$SNP,
outcome = "ieu-b-2"
)
View(outcome)

